Just over a week ago the long-awaited AlphaFold2 (AF2) method paper and associated code finally came out, putting to rest questions that I and many others raised about public disclosure of AF2. Already, the code is being pushed in all sorts of interesting ways, and three days ago the companion paper and database were published, where AF2 was applied to the human proteome and 20 other model organisms. All in all I am very happy with how DeepMind handled this. I reviewed the papers and had some chance to mull over the AF2 model architecture during the past couple of months (it was humorous to see people suggest that the open sourcing of AF2 was in response to RoseTTAFold—it was in fact DeepMind’s plan well before RoseTTAFold was preprinted.) In this post I will summarize my main takeaways about what makes AF2 interesting or surprising. This post is not a high-level summary of AF2—for that I suggest reading the main text of the paper, which is a well-written high-level summary, or this blog post by Carlos Outeiral. In fact, I suggest that you read the paper, including the supplementary information (SI), before reading this post, as I am going to assume familiarity with the model. My focus here is really on technical aspects of the architecture, with an eye toward generalizable lessons that can be applied to other molecular problems.
Continue readingCategory Archives: Biology
AlphaFold2 @ CASP14: “It feels like one’s child has left home.”
The past week was a momentous occasion for protein structure prediction, structural biology at large, and in due time, may prove to be so for the whole of life sciences. CASP14, the conference for the biennial competition for the prediction of protein structure from sequence, took place virtually over multiple remote working platforms. DeepMind, Google’s premier AI research group, entered the competition as they did the previous time, when they upended expectations of what an industrial research lab can do. The outcome this time was very, very different however. At CASP13 DeepMind made an impressive showing with AlphaFold but was ultimately within the bounds of the usual expectations of academic progress, albeit at an accelerated rate. At CASP14 DeepMind produced an advance so thorough it compelled CASP organizers to declare the protein structure prediction problem for single protein chains to be solved. In my read of most CASP14 attendees (virtual as it was), I sense that this was the conclusion of the majority. It certainly is my conclusion as well.
Continue readingThe Future of Protein Science will not be Supervised
But it may well be semi-supervised.
For some time now I have thought that building a latent representation of protein sequence space is a really good idea, both because we have far more sequences than any form of labelled data, and because, once built, such a representation can inform a broad range of downstream tasks. This is why I jumped at the opportunity last year when Surge Biswas, from the Church Lab, approached me about collaborating on exactly such a project. Last week we posted a preprint on bioRxiv describing this effort. It was led by Ethan Alley, Grigory Khimulya, and Surge. All I did was to enthusiastically cheer them on, and so the bulk of the credit goes to them and George Church for his mentorship.
Continue readingAlphaFold @ CASP13: “What just happened?”
Update: An updated version of this blogpost was published as a (peer-reviewed) Letter to the Editor at Bioinformatics, sans the “sociology” commentary.
I just came back from CASP13, the biennial assessment of protein structure prediction methods (I previously blogged about CASP10.) I participated in a panel on deep learning methods in protein structure prediction, as well as a predictor (more on that later.) If you keep tabs on science news, you may have heard that DeepMind’s debut went rather well. So well in fact that not only did they take first place, but put a comfortable distance between them and the second place predictor (the Zhang group) in the free modeling (FM) category, which focuses on modeling novel protein folds. Is the news real or overhyped? What is AlphaFold’s key methodological advance, and does it represent a fundamentally new approach? Is DeepMind forthcoming in sharing the details? And what was the community’s reaction? I will summarize my thoughts on these questions and more below. At the end I will also briefly discuss how RGNs, my end-to-end differentiable model for structure prediction, did on CASP13.
Continue readingProtein Linguistics
For over a decade now I have been working, essentially off the grid, on protein folding. I started thinking about the problem during my undergraduate years and actively working on it from the very beginning of grad school. For about four years, during the late 2000s, I pursued a radically different approach (to what was current then and now) based on ideas from Bayesian nonparametrics. Despite spending a significant fraction of my Ph.D. time on the problem, I made no publishable progress, and ultimately abandoned the approach. When deep learning began to make noise in the machine learning community around 2010, I started thinking about reformulating the core hypothesis underlying my Bayesian nonparametrics approach in a manner that can be cast as end-to-end differentiable, to utilize the emerging machinery of deep learning. Today I am finally ready to start talking about this long journey, beginning with a preprint that went live on bioRxiv yesterday.
The Quantified Anatomy of a Paper
I previously blogged on my adventures in self quantification (QS). In that post I wrote about the general system but did not delve into specific projects. Ultimately however the utility of self quantification is in the detailed insights it gives, and so I’m going to dive deeper into a project that passed a major milestone earlier today: publication of a paper. If you’re interested in the science behind this project, see my other post, A New Way to Read the Genome. Here I will focus on the application and utility of QS as applied to individual projects.
Continue reading
A New Way to Read the Genome
I am pleased to announce that earlier today the embargo was lifted on our most recent paper. This work represents the culmination of over two years of effort by my collaborators and I. You can find the official version on the Nature Genetics website here, and the freely available ReadCube version here. In this post, I will focus on making the science accessible to the lay reader. I have also written another post, The Quantified Anatomy of a Paper, which delves into the quantified-self analytics of this project.
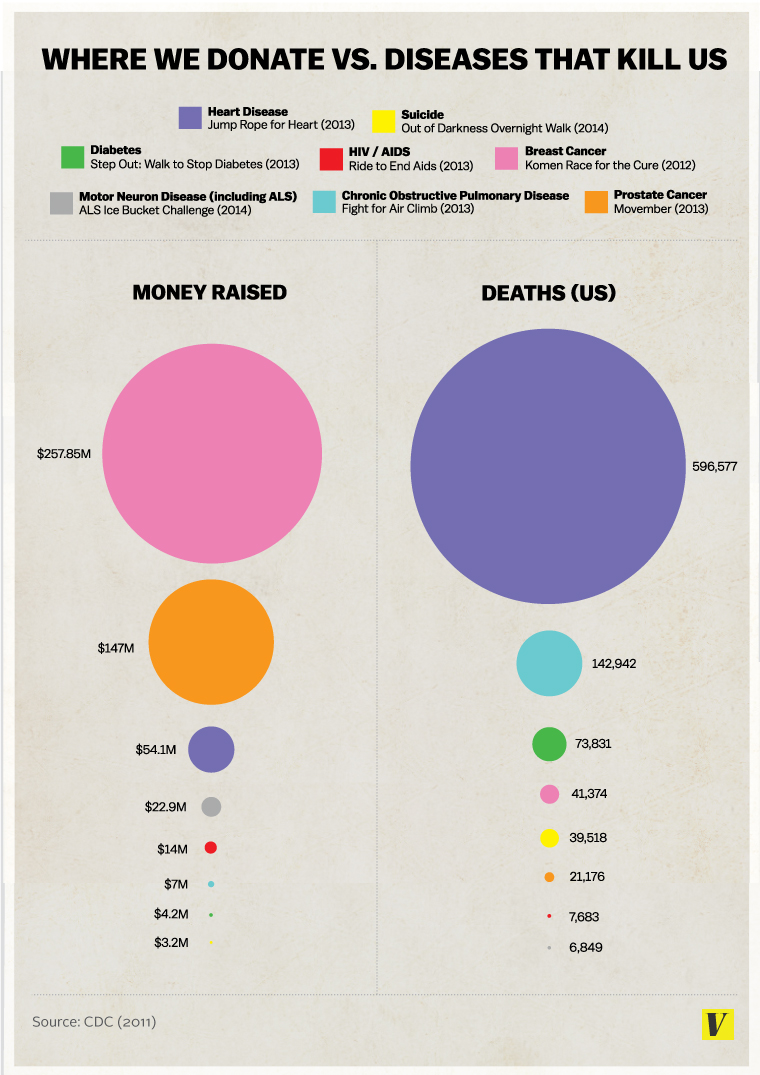
NIH Spending Versus Diseases That Kill Us
An infographic has been making the rounds lately, purporting to depict the amount of money donated to help fight various diseases versus the number of actual deaths caused by each disease. This is the original infographic:

Predictions Are Cheap in Biology
I just came back from ICSB 2013, the leading international conference on systems biology (short write-up here). During the conference Bernhard Palsson gave a great talk, which he ended by promoting a view that (I suspect) is widely held among computational and theoretical biologists but rarely vocalized: most high-impact journals require that novel predictions are experimentally validated before they are deemed worthy for publication, by which point they cease to be novel predictions. Why not allow scientists to publish predictions by themselves?
ICSB 2013
I recently had the pleasure of attending the 14th International Conference on Systems Biology in Copenhagen. It was a five-day, multi-track bonanza, a strong sign of the field’s continued vibrancy. The keynotes were generally excellent, and while I cannot help but feel a little dismayed by the incrementalism that is inherent to scientific research and that is on display in conferences, the forest view was encouraging and hopeful. This is one of the most exciting fields of science today.

{kind=link}